引言
单细胞RNA测序(scRNA-Seq)技术被广泛应用于生产在单细胞水平上的高质量转录组学数据。这些强大的高分辨率数据使得研究人员能够评估各种扰动(如药物反应、CRISPR等)对于不同细胞的影响、探索发育的时间-空间过程、构建参考单细胞图谱,并进行在细胞水平上的数据分析。更进一步地,利用单细胞RNA或DNA测序,各种工作已经能够扩展到基因表达之外的其他模式,如染色质可及性和空间信息等。然而,如何避免对结果的误解、从大量数据中发现有意义的结论,依赖的不仅是关键的技术策略和分析经验,也需要高复杂度的计算机工具辅助。然而,如何创建具有充分功能同时又可获取有效生物学见解的工具是复杂且困难的课题。
方法体系
经典机器学习方法
主成分分析
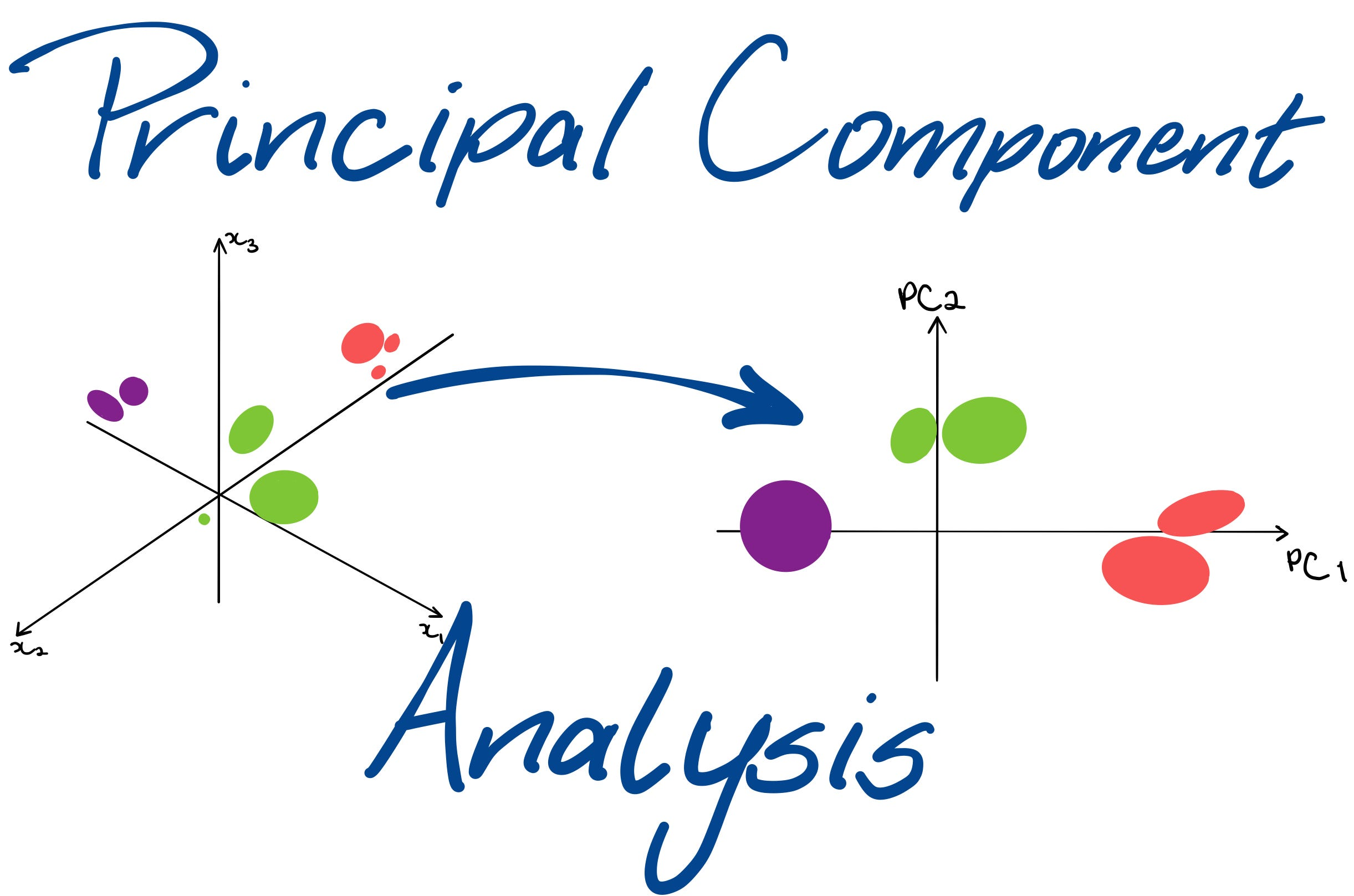
主成分分析(Principal Component Analysis, PCA)是转录组学中最常用的降维方法,通过线性变换将高维基因表达数据投影到低维空间,保留最大方差方向,进而降维并保留关键信息。
了解更多非负矩阵分解
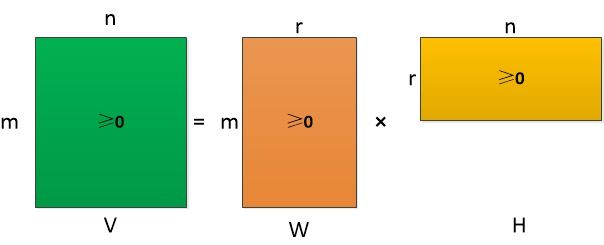
非负矩阵分解(Non-negative Matrix Factorization, NMF)是一种利用优化方法将复杂数据分解为非负组分矩阵的方法,在单细胞RNA测序中常用于对细胞表达情况进行分解,可识别基因共表达模块和细胞亚群特征,提供生物学可解释的特征提取。
了解更多经典机器学习方法在转录组学分析中有着广泛应用。在单细胞RNA测序数据分析中,这些方法通常用于降维、聚类和分类任务。降维技术如主成分分析(PCA)和t-分布随机邻域嵌入(t-SNE)能够将高维基因表达数据映射到低维空间,便于可视化和后续分析。无监督聚类算法如K-means和层次聚类帮助研究人员识别细胞亚群,而监督学习方法如随机森林和支持向量机则用于预测细胞类型或表型特征。这些经典方法的优势在于计算效率高、理论基础扎实,并且在解读和调整模型参数方面相对透明。然而,它们在处理高噪声、高稀疏度的单细胞数据时面临挑战,难以捕捉数据中的复杂非线性关系和生物学变异。尽管如此,这些方法仍然是转录组学分析的基础工具,并经常与更先进的深度学习方法结合使用,以提供更全面的数据解读。
其中,非负矩阵分解方法备受关注。非负矩阵分解(NMF)是一种无监督学习方法,旨在将高维数据矩阵分解为两个非负矩阵的乘积。它在单细胞RNA测序数据分析中被广泛应用,尤其是在细胞类型鉴定和基因表达模式识别方面。NMF的优势在于其可解释性和生物学相关性,能够揭示潜在的基因表达模式和细胞亚群。通过对基因表达数据进行NMF分解,可以识别出具有相似表达特征的基因集合,从而帮助研究人员理解细胞的功能状态和生物学过程。
解纠缠表征学习
Unsupervised Deep Disentangled Representation of Single-Cell Omics
Towards Causal Representation Learning
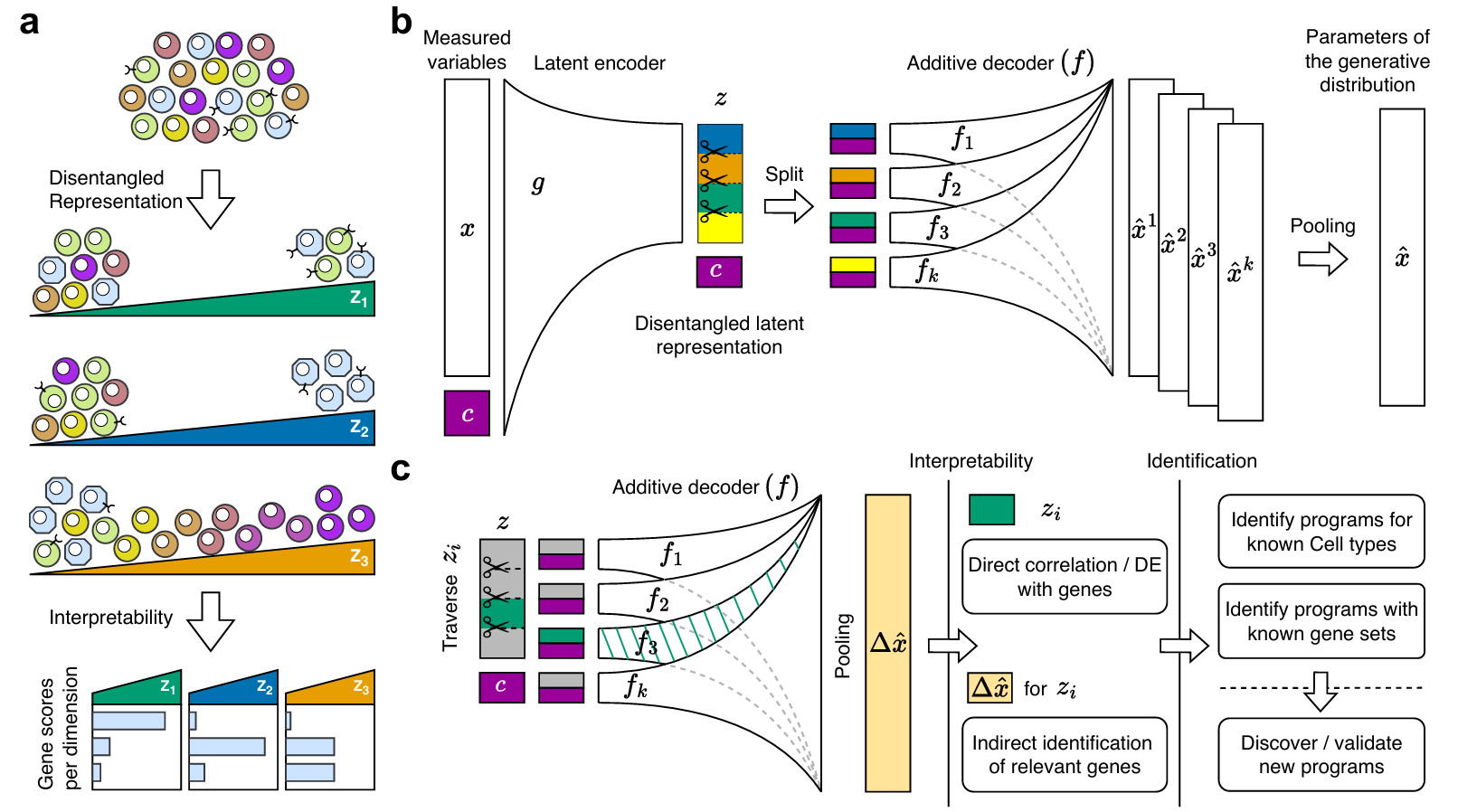
由神经网络等机器学习方法生成高维数据的低维表示这一过程被称为表征学习。表征学习领域作为机器学习的核心领域,已有许多研究旨在增强其表现性能,解纠缠是其重要研究方向之一。解纠缠(Disentangle)是指模型在单个隐空间维度中分离和表示不同隐含的过程或成分的能力。当前在单细胞组学数据上已经有几种隐空间嵌入模型声称能够提供解纠缠的隐空间表示;然而,它们要么依赖于线性假设或强加稀疏性限制,要么依赖于用于监督分离的细胞元数据,进而实现强制的解纠缠。在实践中,并非对于所有数据都有可用的监督数据,尤其是在人为设计的扰动或创新性研究中更是如此。通常认为线性模型无法完全捕获生物学数据的复杂性,尤其对于多个批次、大量细胞、高特征维度的单细胞组学数据,线性模型不能充分地产生跨批次、跨组织、跨细胞类型的全局特征。正因为需要克服这些问题,具有无监督、非线性特征的解纠缠表征学习在单细胞组学领域具备很大的潜力。解纠缠表征学习(Disentangled Representation Learning, DRL)作为因果深度学习与表征学习的交叉应用,旨在将数据中的复杂因素分离为独立的、语义清晰的表征,减少不同表征维度的冗余和依赖性。这种表征可以提升模型的泛化能力、提高表征维度的可解释性,并增强对未见数据的鲁棒性。
单细胞组学上的Transformer架构
scGPT: toward building a foundation model for single-cell multi-omics using generative AI
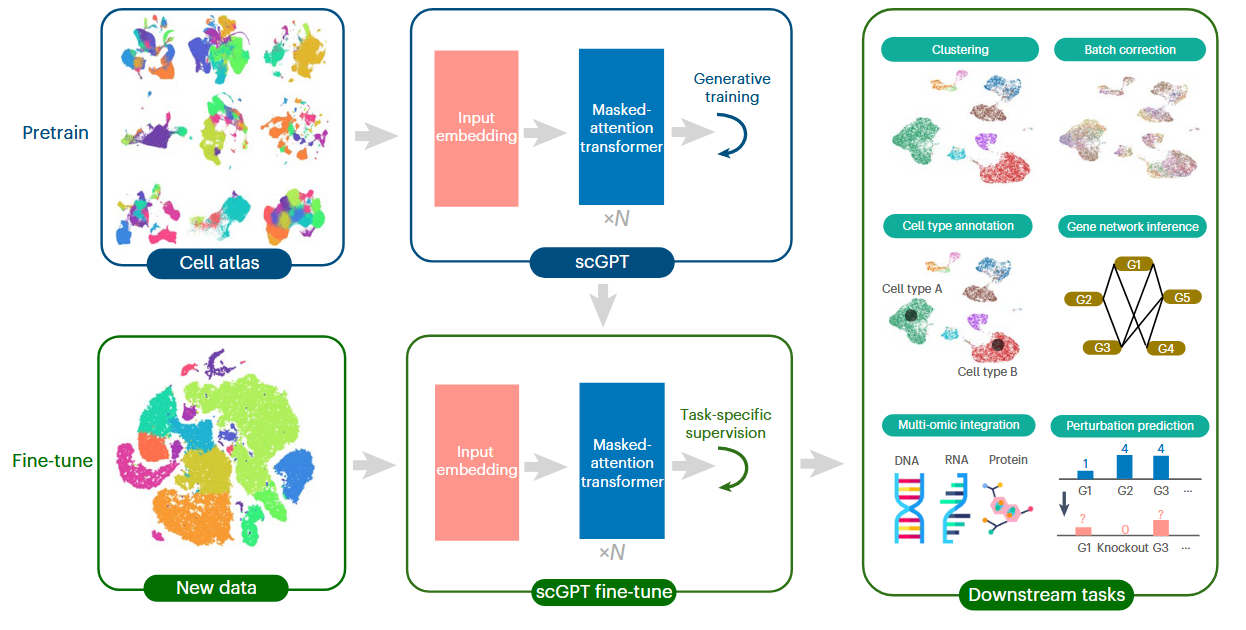
尝试构建了单细胞生物学的类GPT式预训练基础模型scGPT,基于超过3300万个细胞表达数据上进行预训练,在多个下游任务上取得了卓越的性能。
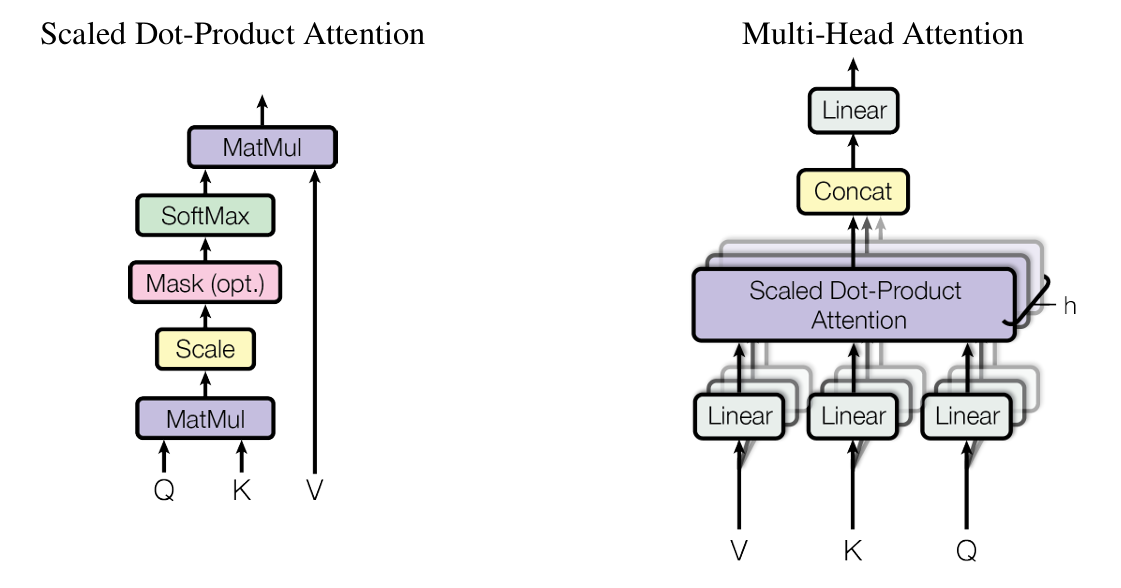
了解更多单细胞组学的出现提供了对细胞异质性和动态过程的精细识别,但其也是一种挑战:目前的分析方法在捕获各种大规模单细胞数据集的方面存在不足。与此同时,深度学习领域见证了Transformer模型带来的巨大变革。Transformer模型的一个主要特征是其结构中引入了一组特殊的序列运算,称之为注意力机制。注意力机制通过定义元素之间的相互运算产生注意力权重,通过注意力权重构建有向图,允许不同序列元素间产生信息的定向流动。这种注意力权重可以被模型输出以分析模型的行为,这为Transformer模型带来了优越的可解释性,进而有许多工作关注于将该模型结构引入单细胞组学深度学习中。 以往的大多数深度学习模型将输入视为一个向量(在单细胞组学的背景下输入可以代表一个细胞,此时向量的每个元素代表一个基因的RNA表达量),并迭代地应用一系列的可学习的非线性变换,最终将输入变换为所需的数值。单细胞应用中的常见模型架构是不依赖于数据注释即可训练的自编码器(Autoencoder),该架构由编码器和解码器组成,训练要求其能够通过编码器将输入映射为低维的隐空间表示,而解码器能将隐空间表示准确地重建回输入。隐空间的维度瓶颈迫使模型学习一个有效的特征提取器,捕获全局上的模式并过滤掉噪声,仅保留导致细胞产生主要变化的有意义的隐空间表示。Transformer架构则使用注意力机制来处理表示为一组高维向量嵌入的输入数据,根据数据样本中所有输入特征来调整他们处理某一特定输入特征的方式,在单细胞组学中,这允许Transformer根据当前正在处理的细胞类型灵活地解释不同的基因作用模式,同时为Transformer模型赋予了强大的解释性,可以通过注意力矩阵直接得到基因间潜在的相关性。
当前在单细胞转录组学领域,Transformer结构模型往往被用于构建大型预训练模型,旨在通过学习并整合大量、多模态的单细胞组学数据,产生对于细胞的立体认知,进而完成一系列有关于细胞状态的任务(例如细胞扰动预测、细胞标签整合和预测等)。尽管大型预训练模型往往具备更优越的任务性能,但仍然会由于不同实验产生的数据中严重的技术伪影、批次效应和噪声的问题导致无法进行泛化学习。另一方面,由于引入了不必要的计算量和结构限制且无法取得优越性能,使用Transformer结构构建即插即用式的组学分析模型的探索较少。一篇相关的研究利用该结构实现在细胞转录组数据上构建分类器的任务,并给出了利用注意力权重解读分类依据的常规思路,取得了较好的可解释性突破。这种可解释性如果能用于细胞表达程序解析,将解决深度学习方法难以产生生物学见解的难题。
典型应用:肿瘤免疫表达程序分析
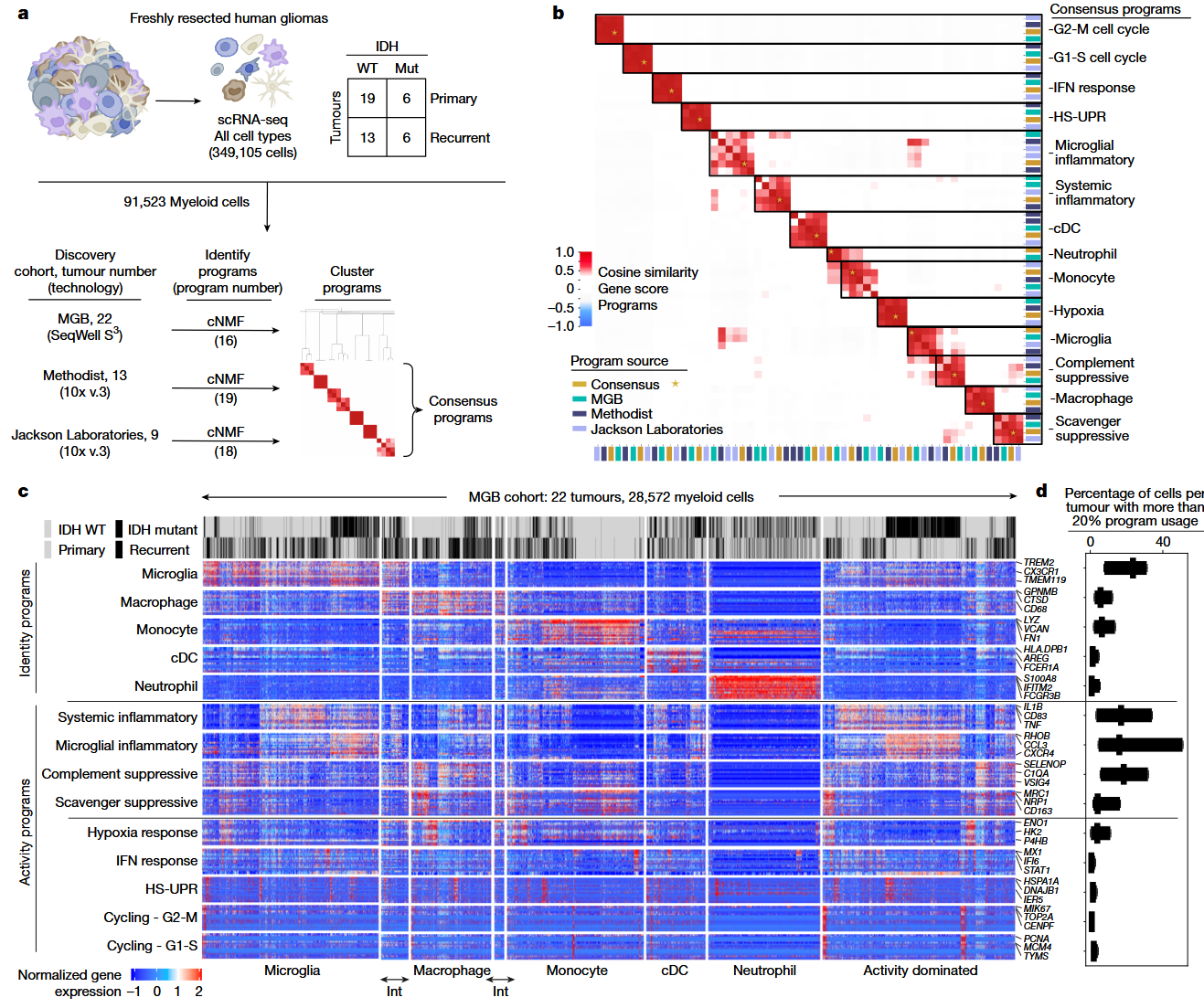
一项发表于Nature的研究充分利用了可解释机器学习解纠缠方法共有非负矩阵分解(consensus Non-negative Matrix Factorization, cNMF)1对神经胶质瘤中复杂的髓系细胞进行了表达程序分解的研究2。研究通过整合单细胞RNA测序、染色质可及性、空间转录组学和神经胶质瘤类器官外植体系统来系统地研究其表型,并利用cNMF鉴定并发现了四种免疫调节表达程序,其中有两种原发性脑肿瘤中特有的:小胶质细胞炎症反应和清道夫免疫抑制程序;以及广泛存在于各类免疫中的系统性炎症和补体免疫抑制程序,它们也由非脑肿瘤表达。这些表达程序不取决于髓系细胞类型、发育起源或肿瘤突变状态,而是由微环境线索驱动,包括肿瘤缺氧情况、白细胞介素-1β、TGFβ 和地塞米松治疗等。它们的相对表达可以预测免疫治疗反应和总生存期。通过将相应的程序与介导基因组元件、转录因子和信号通路相关联,研究发现了操纵髓系细胞表型的策略,进而为开发更有效的免疫疗法奠定了基础。
挑战与对策
可解释性与性能的平衡
可解释机器学习方法在转录组学分析中面临的一个主要挑战是如何在可解释性和模型性能之间取得平衡。许多可解释性方法可能会牺牲模型的预测性能,导致结果不够准确。因此,在选择和应用可解释机器学习方法时,需要仔细考虑这一平衡,以确保所得到的结果既具有生物学意义,又能提供可靠的预测。
数据质量与噪声处理
转录组学数据通常受到噪声和批次效应的影响,这可能会干扰可解释机器学习方法的性能。为了提高模型的可靠性,研究人员需要在数据预处理阶段采取适当的去噪和标准化方法,以减少这些影响。此外,使用更强大的模型和算法来处理高噪声数据也是一个重要的研究方向。当前的一个重要研究方向便是如何在去除由于技术批次、实验设计等因素造成的伪影的同时,保留生物学上有意义的信号。为此,研究者们提出了多种方法来处理这些问题,包括使用深度学习模型进行数据去噪、批次效应校正和数据标准化等3。